|
Works Cited:
"Essentials Of Genetics Unit 2: How Does DNA Move From Cell To Cell?". Nature.Com, 2014, https://www.nature.com/scitable/ebooks/essentials-of-genetics-8/126042302#bookContentViewAreaDivID. Accessed 24 Feb 2018. “Mitosis/Meiosis POGIL”
0 Comments
IntroductionPCR, short for Polymerase Chain Reaction, is a method used to essentially copy small segments of DNA. It can be thought of as a “molecular photocopier”. Learning about PCR is important, as it is integral to DNA work because many copies of DNA are needed for molecular and genetic analyses. Materials and MethodsIn this lab, I worked with Olivia Hicks. Her blog can be found here. The first thing we did was pour the gel we would be using in the lab. We shared the gel with Olivia Studebaker, Tejal Patel, Andrew Draheim, and Nathan Washecheck. Next, we slipped on some gloves and got to the real exciting work. We put gloves on because we did not want to contaminate our reaction with our own DNA or any oils that exist on our skin. In this lab, every group that shared a gel had a control tube, which for us was prepared by Nathan and Andrew. The control tube had the same composition of the other tubes, but was not run through the thermocycler. Olivia and I prepared our tubes as follows: First, we were given our PCR tubes which we then labeled in a way that we could tell them apart. Each tube contained a little white bead, reminiscent of a Styrofoam bead, that contained the taq polymerase, nucleotides, and salt that we would need for the reaction. After labeling our tubes, we placed them in an ice bath to keep them cold. We then took them out and ensured the beads were at the bottom of the tubes by tapping them on the counter. Next, we added 20 µL of DNA primer mixture to the tubes using our trusty micropipettors and tapped them again to make sure that the primer went to the bottom of the tube. We then added 10 µL of Lambda DNA to our tube using the micropipettors and tapped our tubes once more to ensure that they had been fully mixed. The tubes were then closed and put back onto the ice, while the control was instead put next to the thermocycler. Our group then put the tubes into the thermocycler for Dr. Shingleton to run through the denature-anneal-extend cycles to prepare for the next class period. The next day, we added 4 µL of loading dye/visualization stain to our tubes so that we could be able to see our DNA under the black light. DNA doesn’t naturally glow under UV light, so the dye allowed us to see how far the DNA traveled in the lanes and analyze the results. After adding the dye, we tapped our tubes one last time to mix the contents well, and loaded each lane with about 15 µL of sample from each tube. The control, however, only used 12 µL. 15 µL of standard was also loaded into the gel. After loading, we ran the gel at 110V at 09:46. Since the loading dye had to move 45 mm to get the best results, we boosted the voltage to 120V at 10:16 and ended at around 10:20. After carefully removing our gel, (if you have read my last lab, you might remember that the gels cannot survive gravity induced trauma) we placed it in the UV light box to analyze our results. ResultsConclusionPCR is a technique used in DNA labs to essentially multiply the amount of DNA that is available for analysis. PCR is useful because it is a relatively inexpensive way to increase the amount of useful DNA needed for analysis. This lab was important to our genomics class because it taught us how to use PCR to amplify DNA and analyze it. In this lab, I learned about the process of PCR, and the importance of the thermocycler as well. As you might remember, the control was not put into the thermocycler during our experiment. As a result, we saw that the control sample only traveled 26mm as opposed to the average of 62.83 of the 6 PCR samples. This showed us that the thermocycler was integral to the process of PCR. Without the thermocycler, the reaction didn’t take place properly in the control tube. During our procedure, Olivia and I didn’t seem to make any errors on our part. However, one aspect I would like to change was the duration of the lab. It seems to me that the lab could have taken only a few hours if we were able to do it in one go. Instead, due to our school schedule, we had to complete the lab over several days, increasing the chance of contamination and error in the process. All in all, the lab was probably my favorite so far, as I felt like I was doing real DNA analysis, as opposed to the previous lab we did where we analyzed M&M dyes. One question I had while I was doing this lab was whether PCR is used in the process of cloning animals. For example, if there was only a small sample of a Wooly Mammoth’s DNA available for analysis, could PCR be used to amplify that DNA in order to preserve the original sample and allow for multiple trials? DiscussionIn the PCR technique, the DNA sample to be amplified is combined with a primer and is denatured, or separated into two pieces of single stranded DNA, through heat. This is because the heat used during PCR breaks the hydrogen bonds between the two strands of DNA in the double helix. After this, the taq polymerase enzyme builds two new strands of DNA using the original strands as a template. As a results, the new molecules of DNA that are created contain one old and one new strand of DNA. The thermocycler directs the process of denaturing and synthesis by altering the temperature every few minutes, up to 30 or 40 times to create more than 1 billion copies of the original DNA segment. Overall, PCR is a nifty technique that creates many copies of a sample of DNA, of which can be used for analysis. While DNA replication in a cell and DNA amplification by PCR are similar in the fact that they create duplicate strands of DNA, both break hydrogen bonds in the beginning of the process, and both uses primers, it is important to note the key differences between the two. DNA replication takes place in a cell at a much lower temperature than PCR. In replication, the DNA helicase enzyme “unzips” the strand of DNA to begin the process. In PCR, heat is used to break the hydrogen bonds between the strands since DNA helicase is not easily reproduced. In DNA replication, an RNA primer is used to give polymerase something to bond to. Much like in replication, a primer is used in PCR, however a DNA primer is used as opposed to an RNA primer. In replication, an RNA primer is used because the cell doesn’t have an extra DNA to use. Lastly, PCR uses taq polymerase, an enzyme found in boiling mud pits. Taq polymerase is used in PCR due to the high temperatures (up to 94°C) at which the DNA denatures and the synthesis must occur. Using polymerase from our own human body wouldn’t be feasible because the temperature would be well out of range for the functioning temperature of the enzyme. Before we ran our gels, we added a stain to our sample in order to see it. This is because DNA doesn’t glow under UV light naturally. In order to properly analyze our DNA, we need to see how far it traveled. Since the DNA we used was clear, it would have been impossible to see how far the samples traveled using natural light. We added to stain so that we could see how far the DNA traveled. Using the stain to analyze our DNA, Olivia and I estimated the size of the amplicon to be 1,400. The known size was 1,106. Our percent error of 21% was probably due to the fact that it was very difficult to see exactly where each band ended. This experiment greatly increased my knowledge of PCR and makes me exciting for the experiments to come! Bonus Works Cited“Polymerase Chain Reaction (PCR) Fact Sheet.” National Human Genome Research Institute (NHGRI), www.genome.gov/10000207/polymerase-chain-reaction-pcr-fact-sheet/.
DNA, short for deoxyribonucleic acid is an extremely complex molecule that contains all the information necessary to build and maintain an organism. It serves to pass on genetic information from parent to offspring, a characteristic known as heredity. DNA is composed of smaller subunits called nucleotides, of which there are four varieties. Nucleotides are made up of three main components, a 5-carbon sugar molecule called deoxyribose, a phosphate group, and a nitrogenous base. The nitrogenous base is responsible for the identity of the nucleotide. DNA is composed of four types of nucleotides: adenine (A), cytosine (C), guanine (G), and thymine (T). These nucleotides bond in pairs, with A and T only bonding to each other, and G and C bonding to each other. In total, there are over 246 million nucleotides in one chromosome, and over 6 billion in one cell. The picture below shows the structure of DNA in detail. In order for DNA to be passed on from parent to offspring, it must be replicated. 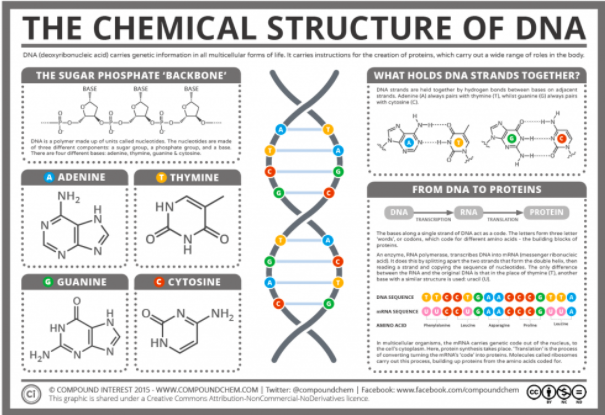  The process of DNA replication starts with helicase, an enzyme that pulls apart the double stranded DNA. It stars in an area rich with A-T pairs since these are easier to separate because they contain two hydrogen bonds as opposed to C-G pairs that contain three. Once the double stranded structure is split, two DNA polymerase enzymes collaborate to copy the leading strand and the lagging strand. On the leading strand, DNA polymerase binds the nucleotides in 5’-3’ direction, while RNA primase inserts starter RNA primer at the initial point, giving the DNA polymerase a signal to start adding the corresponding nucleotides to the strand. On the lagging strand however, RNA starts the binding process. DNA polymerase has to work backwards in 3’-5’ direction, resulting in okazaki fragments, which are short DNA segments that make up the lagging strand of the newly synthesized DNA. After finishing one okazaki fragment, the “clamp” that secures DNA polymerase to the lagging strand dissociates and lets DNA polymerase release the lagging strand temporarily. As the double stranded structure keeps getting split by the helicase, the RNA primase is initiated and inserts a short RNA primer. Then DNA polymerase clamps back to the lagging strand again and begins working from where the primers are. The polymerase stops when it reaches the point where the preceding okazaki fragment is and releases the lagging strand, waiting for RNA primase to signal. Once the copying work is done, an enzyme called exonuclease takes away the RNA primer, and DNA polymerase replaces the gap with DNA nucleotides. At the end of the process, ligase fill the gaps left in the sugar-phosphate backbone.  The structure of DNA easily facilitates the function of replication. Despite the strong double helix that stabilizes the base pairs inside the structure, the weak hydrogen bonds that hold the base pairs together allow the molecule to untwist easily for replication. The DNA molecule is split down the middle into two strands and is now able to create two copies of DNA. Various enzymes are present to stimulate the reaction as the base pairs dislodge from each other. Since the nucleotides are exposed, corresponding base pairs match up to recreate the ladder structure. Once this is complete, this newly generated DNA strand coils back up into the double helix. DNA replication is a crucial part of cell reproduction in all living organisms. In fact, life is dependent on this because without replicating DNA, our information would not pass down to future generations and life would no longer exist. Works Cited:
|
AuthorAllan Kalapura. Holland Hall class of 2019. Archives |
 RSS Feed
RSS Feed
Photo used under Creative Commons from Kevin M. Gill
